Подробнейший разбор текущего понимания коагулопатии, связанной с COVID-19, опубликовал научный медицинский журнал NatureReviewsImmunology. Помимо обобщения ключевых идей, международная команда экспертов, представляющих ведущие медицинские факультеты и университеты США, Канады, Нидерландов, обозначила основные пробелы в текущих знаниях, наметив своего рода дорожную карту для будущих исследований. Предлагаем ознакомиться с основными положениями этой важной работы.
Значимость проблемы
COVID-19-ассоциированная коагулопатия (CAC) является опасным для жизни осложнением коронавирусной инфекции SARS-CoV-2. Однако клеточные и молекулярные механизмы, лежащие в основе этого состояния, остаются неясными. Понимание патогенеза на геномном, молекулярном и клеточном уровнях необходимо для снижения угрозы развития тромбоза у пациентов группы риска. Существующая на данный момент концепция САС включает три основных патологических механизма: дисфункцию эндотелиальных клеток сосудов, гипервоспалительный иммунный ответ и гиперкоагуляцию.
По сравнению с заболеваниями, вызванными другими распространенными респираторными вирусными инфекциями, у пациентов с COVID-19 отмечается более высокая частота и тяжесть тромбозов.
В недавнем отчете о клиническом исследовании ВОЗ ACTIV-4B (Effect of Antithrombotic Therapy on Clinical Outcomes in Outpatients With Clinically Stable Symptomatic COVID-19) предполагается, что уровень D-димера повышен примерно у 10 % всех пациентов с COVID-19, что указывает на высокую частоту коагулопатии.
Этиопатогенез COVID-19-ассоциированной коагулопатии не соответствует классическому диссеминированному внутрисосудистому свертыванию крови. Найдены доказательства того, что САС предполагает сложные дисрегулированные взаимодействия между воспалительной, иммунной, коагуляционной, фибринолитической, комплементарной и калликреин-кининовой системами. Эта связь врожденного иммунного ответа со свертыванием крови — в частности, в микрососудах, где дисфункция эндотелиальных клеток способствует образованию тромбов и воспалению, — была названа «иммунотромбозом».
Это развивающаяся концепция, предполагающая сложные взаимоотношения между циркулирующими иммунными клетками, эндотелием сосудов и множеством растворимых и мембраносвязанных факторов, регулирующих коагуляцию, врожденный и адаптивный иммунитет. Эта парадигма все больше внедряется в понимание тромбоза в различных условиях, включая рак, сепсис и острый респираторный дистресс-синдром. Однако в контексте COVID-19 до сих пор не хватает детального понимания молекулярных и клеточных взаимодействий каждой из этих систем. Поэтому так важно рассмотреть все три пути, которые становятся ключевыми патологическими механизмами при САС: дисфункция эндотелиальных клеток сосудов, гипервоспалительный иммунный ответ и гиперкоагуляция.
Дисфункция эндотелиальных клеток сосудов при COVID-19
В нормальных условиях эндотелий сосудов обеспечивает антитромботическое состояние поверхности, экспрессируя молекулы, которые предотвращают активацию тромбоцитов (например, оксид азота, простациклин и эктонуклеотидазы) и коагуляцию (например, ингибитор пути тканевого фактора (TFPI), тромбомодулин и рецептор протеина С эндотелиальных клеток (EPCR)).
Эндотелий сосудов также выстлан богатым углеводами гликокаликсом, который включает в себя антикоагулянтные молекулы, такие как гепарансульфат, и служит для защиты от вторжения патогенов. Эти тонко регулируемые антитромботические системы могут быть нарушены в условиях бактериальных и вирусных инфекций, при этом происходит функциональная потеря антикоагулянтных молекул, таких как TFPI, тромбомодулин и EPCR, с поверхности эндотелия, повышается экспрессия прокоагулянтов (в первую очередь тканевого фактора) и повреждение гликокаликса.
Считается, что при инфекции SARS-CoV-2 дисфункция эндотелия сосудов вносит основной вклад в патогенез васкулопатии COVID-19. На ранних стадиях заболевания это происходит в основном в легких, где SARS-CoV-2 посредством ангиотензин-превращающего фермента 2 (ACE2) инфицирует пневмоциты 2-го типа, клетки, которые находятся в непосредственной близости от альвеолярной сосудистой сети. Повреждение альвеол и эндотелия микрососудов приводит к усилению воспаления, инфильтрации активированных нейтрофилов, высвобождению нейтрофильных внеклеточных ловушек (NETs), активации комплемента и диффузному тромбозу микрососудов, что способствует нарушению дыхательной функции и ухудшению прогноза для пациента.
Пока неизвестно, вызвано ли повреждение эндотелия вначале инфицированием SARS-CoV-2 напрямую, косвенно, в результате так называемого «цитокинового шторма», либо другими врожденными, адаптивными либо аутоиммунными явлениями. Однако по мере прогрессирования заболевания, вероятно, множество факторов вносят свой вклад в тяжесть поражения эндотелия, степень тромбоза и повреждения органов. Выяснение конкретных триггеров и факторов, определяющих тяжесть заболевания, все еще находится в стадии исследования.
Дисрегуляция калликреин-кининовой системы и повреждение эндотелиальных клеток
Хотя дисфункция эндотелия сосудов может возникнуть при любой тяжелой инфекции, SARS-CoV-2 обладает уникальным взаимодействием с рецептором ACE2 на поверхности клеток хозяина, что может вызвать дисфункцию эндотелиальных клеток на ранней стадии инфекции. SARS-CoV-2 связывается с ACE2, присутствующими на эпителиальных клетках легких, вызывая эндоцитоз комплекса SARS-CoV-2 – ACE2, следовательно, уровень экспрессии АСЕ2 на поверхности клеток падает в месте инфекции, что приводит к локальному дефициту АСЕ2.
АСЕ2 выполняет двойную функцию, регулируя как ренин-ангиотензин-альдостероновую, так и калликреин-кининовую системы. Кинины — вазоактивные пептиды, которые вызывают расслабление сосудистых гладкомышечных клеток и увеличение проницаемости сосудов. Дисрегуляция деградации кининов, вызванная прямым воздействием вируса на ACE2, может увеличить активность кининов, что способствует развитию ангионевротического отека. Таким образом, калликреин-кининовая система может быть активирована системно в процессе COVID-19, что приводит к «цитокиновому шторму», вызывающему повышенную проницаемость сосудов, воспаление, накопление жидкости и повреждение органов.
Заметно повышенный уровень D-димеров, наблюдаемый при тяжелой форме COVID-19, указывает на то, что отложение фибрина и последующая его деградация играют роль в патофизиологии COVID-19. Однако относительный вклад внутри- и внесосудистого образования и лизиса фибрина в D-димеры неясен. Расщепление плазмином этого внесосудистого фибрина и утечка D-димеров в кровь могут способствовать повышенному уровню D-димеров у пациентов с COVID-19.
Действительно, во время инфекции SARS-CoV-2 протромботические TF и внеклеточные везикулы высвобождаются из множества типов клеток. Преобладающим источником TF-положительных (TF+) внеклеточных везикул, обнаруженных в бронхоальвеолярной лаважной жидкости (БАЛЖ) пациентов с COVID-19, вероятно, являются активированные эпителиальные клетки легких. Хотя клеточное происхождение внеклеточных везикул TF+ в крови окончательно неизвестно, они, вероятно, в основном образуются из активированных моноцитов, в их образовании также участвуют эндотелиальные клетки, нейтрофилы и эпителиальные клетки. Независимо от источника, повышенный уровень циркулирующих внеклеточных везикул TF+ связан с увеличением тяжести заболевания, включая повышенную частоту венозной тромбоэмболии у пациентов с COVID-19, что убедительно свидетельствует об их участии в CАС.
SARS-CoV-2 и гематоэнцефалический барьер
Помимо легких, клинически важными мишенями SARS-CoV-2 являются почки, печень, сердце, кожа и центральная нервная система, особенно головной мозг (см. рис. 1).
Рисунок 1. Потенциальные клинические проявления COVID-19-ассоциированной коагулопатии.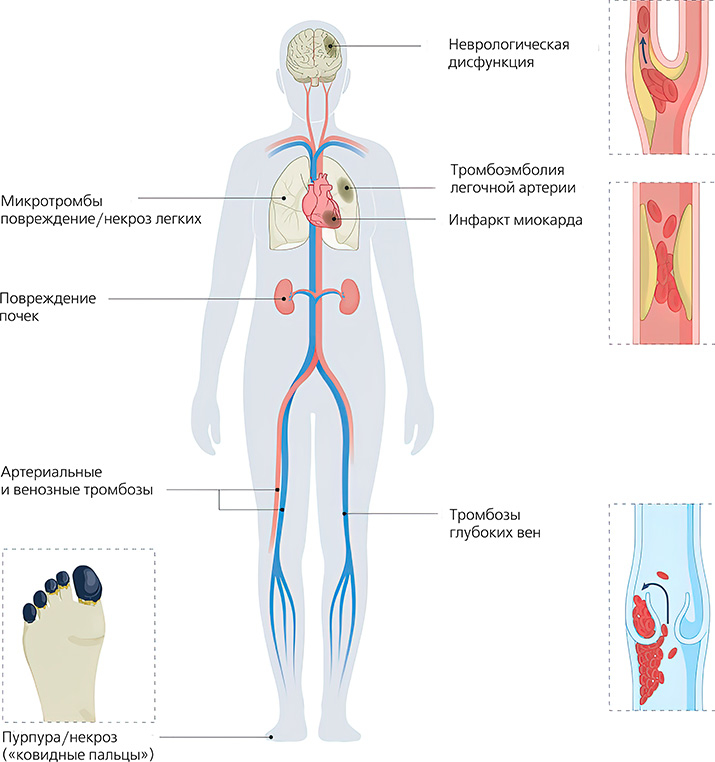 COVID-19-ассоциированная коагулопатия характеризуется нарушениями коагуляции, которые затрагивают множество тканей и органов и варьируют от кожной пурпуры (также известной как «ковидные пальцы») до инфаркта миокарда и неврологической дисфункции. Циркулирующие микро- и/или макротромбы могут привести к повреждению нескольких органов одновременно.
COVID-19-ассоциированная коагулопатия характеризуется нарушениями коагуляции, которые затрагивают множество тканей и органов и варьируют от кожной пурпуры (также известной как «ковидные пальцы») до инфаркта миокарда и неврологической дисфункции. Циркулирующие микро- и/или макротромбы могут привести к повреждению нескольких органов одновременно.
При серийном гистологическом анализе 100 аутопсий с положительной реакцией COVID-19 в мозге 58 человек были обнаружены широко распространенные микротромбы и микроинфаркты в неокортексе, причем наличие микротромбов часто ассоциировалось с мелкими и точечными инфарктами. Более того, иммуногистохимическое окрашивание выявило высокую экспрессию рецептора АСЕ2 во внутрипаренхимальных кровеносных сосудах.
Исходя из этих наблюдений, неудивительно, что пациенты, инфицированные SARS-CoV-2, часто имеют широкий спектр неврологических симптомов, включая головную боль, нарушения сна, изменения обоняния и вкуса, ишемический инсульт, серьезные когнитивные нарушения, спутанность и снижение уровня сознания. Считается, что эти неврологические проявления инфекции SARS-CoV-2, связанные с повышенным риском внутрибольничной смертности, в первую очередь зависят от иммунотромбоза, что приводит к так называемой дисрегуляции «нейрокоагуляции» с избыточной выработкой тромбина, изменению функции эндотелиальных клеток гематоэнцефалического барьера и нейровоспалению.
Хотя это несколько спорно, потенциально SARS-CoV-2 может инфицировать ткани мозга напрямую, тем самым вызывая повреждение клеток. Однако более вероятным представляется, что вирус также воздействует на мозг через косвенные механизмы. В некоторых посмертных исследованиях пациентов с COVID-19 было обнаружено мультифокальное микрососудистое эндотелиальное повреждение мозга без каких-либо признаков SARS-CoV-2 в мозге, что указывает на косвенное воздействие через защитные механизмы хозяина.
Напротив, в другой серии посмертных исследований РНК SARS-CoV-2 была обнаружена у некоторых пациентов в одной или нескольких областях мозга, что позволяет предположить, что вирус может вызывать прямой нейроцитопатический эффект. Исследования in vitro подтверждают последнюю гипотезу, поскольку SARS-CoV-2 может непосредственно инфицировать нейроны мыши и человека с сопутствующими метаболическими эффектами на инфицированных и соседних нейронах. Однако остается неясным, какие области мозга или типы клеток наиболее восприимчивы к прямому или косвенному повреждению, опосредованному SARS-CoV-2, и как вирус наиболее эффективно проникает в ткани мозга через гематоэнцефалический барьер.
В целом, в настоящее время имеются весомые доказательства того, что инфекция SARS-CoV-2 может вызывать дисфункцию эндотелиальных клеток сосудов и что именно она лежит в основе патологии, наблюдаемой у многих пациентов с тяжелой формой COVID-19. Но есть еще множество научных вопросов, которые только предстоит решить в этой области.
SARS-CoV-2 и сосудистая дисфункция: вопросы, требующие ответов
? Какие механизмы лежат в основе SARS-CoV-2-индуцированной эндотелиальной дисфункции, активации и васкулопатии?
? Как SARS-CoV-2 дифференцированно влияет на сосудистую систему в разных органах в разных местах?
? Каковы последствия нарушения эндотелиального гликокаликса в различных участках сосудистого русла? Какие механизмы лежат в основе нарушения гликокаликса, как его можно контролировать и какие методы лечения могут быть разработаны для смягчения последствий?
? Какую роль в CAC играют циркулирующий тканевой фактор и внеклеточные везикулы, каковы механизмы их высвобождения, можно ли на них воздействовать и являются ли они полезными прогностическими биомаркерами CAC?
? Каковы механизмы прямого и непрямого инфицирования центральной нервной системы, вызванного SARS-CoV-2, и каков патогенез повреждения нейронов?
? Какие особенности эндотелия гематоэнцефалического барьера регулируют проникновение SARS-CoV-2, вирусных белков и медиаторов воспаления в мозг?
Гипервоспалительный иммунный ответ
В посмертных анализах тканей пациентов с COVID-19 постоянно выявляли отложение фрагментов активации комплемента в сосудистой системе многих органов наряду с микро- и макрососудистой эндотелиопатией и тромбозом.
Клинически микрососудистый тромбоз может проявляться в виде кожной пурпуры или состояния, известного как «ковидные пальцы» (см. рис. 1).
Серийные исследования сыворотки крови пациентов с COVID-19 выявили раннюю и стойкую гиперактивацию комплемента с повышенным уровнем циркулирующих протеолитических фрагментов, что является прогностическим фактором неблагоприятного исхода. Эти данные свидетельствуют о том, что SARS-CoV-2 может вызывать активацию комплемента по трем основным путям: классическому, альтернативному и лектиновому.
Дисфункция тромбоцитов, способствующая иммунотромбозу
Хотя тромбоцитопения характерна для многих вирусных заболеваний, она не часто наблюдается при COVID-19. Тем не менее при инфицировании SARS-CoV-2 часто обнаруживается повышенная активация тромбоцитов, вызванная множеством факторов, включая растворимые факторы коагуляции и системы комплемента, воспалительные цитокины, анти-SARS-CoV-2-иммуноглобулины, изменения напряжения сдвига и воздействие активированных эндотелиальных клеток и нейтрофилов. Посредством высвобождения своих внутриклеточных компонентов гранул и экстернализации протромботической мембранной поверхности фосфатидилсерина тромбоциты способствуют запуску и поддержанию иммунотромботической петли усиления, которая способствует развитию САС.
Тромбоциты пациентов с COVID-19 демонстрируют усиленную агрегацию, сопровождаемую повышенной экспрессией P-селектина на поверхности клеток, но с пониженной активацией интегрина αIIbβ3. У пациентов с COVID-19 также повышены циркулирующие уровни растворимых маркеров активации тромбоцитов (например, CC-хемокинового лиганда 5 (CCL5; также известен как RANTES) и тромбоцитарного фактора 4, а также увеличены агрегаты тромбоцитов-нейтрофилов и тромбоцитов-моноцитов.
При повышенной активации эндотелия сосудов эти агрегаты могут легко прилипать к стенке сосуда. Прилипание тромбоцитарно-нейтрофильных агрегатов, вероятно, приводит к высвобождению протромботических и провоспалительных NETs, тогда как агрегаты тромбоцитов-моноцитов могут способствовать гиперкоагуляции через повышенную экспрессию TF моноцитами. В целом убедительны доказательства, указывающие на то, что активированные тромбоциты, нейтрофилы и NETs играют значительную и взаимодополняющую роль в образовании тромба, а также являются предикторами более тяжелого заболевания.
Механизмы, лежащие в основе повышенной реактивности тромбоцитов при COVID-19, по-видимому, включают усиление сигнальных путей MAPK и выработку тромбоксана А2 — ответы, которые, вероятно, связаны с изменениями в транскриптоме тромбоцитов, вызванными SARS-CoV-2.
Анализ РНК-секвенирования, сравнивающий тромбоциты пациентов с COVID-19 с тромбоцитами здорового контроля, выявил более 3 000 дифференциально экспрессированных генов. И только часть этих генов была дифференциально экспрессирована в тромбоцитах пациентов с COVID-19, не находящихся в отделениях интенсивной терапии, по сравнению с поступившими в ОРИТ, что указывает на то, что эти изменения в экспрессии генов тромбоцитов, скорее всего, являются ранним ответом на инфекцию SARS-CoV-2 и не обязательно связаны с тяжестью заболевания.
Неизвестно, происходят ли вызванные SARS-CoV-2 изменения транскриптома тромбоцитов в результате прямой вирусной инфекции тромбоцитов или опосредованно, в результате гипервоспалительной реакции на вирус. Первоначальные исследования показали, что мРНК SARS-CoV-2 может быть обнаружена в тромбоцитах некоторых пациентов с COVID-19. Однако остается неясным, экспрессируют ли тромбоциты ACE2 или существуют другие пути проникновения вируса, например, через внеклеточные везикулы.
Сравнительный анализ транскриптомов тромбоцитов пациентов с COVID-19 с транскриптомами тромбоцитов пациентов с другими вирусными инфекциями позволяет предположить, что, вероятно, существуют альтернативные механизмы проникновения вирусов в клетки хозяина. Например, инфекции вируса денге, гриппа и SARS-CoV-2 характеризуются повышенным уровнем интерферон-индуцируемой трансмембраны 3 (IFITM3), противовирусного белка, критически важного для регулирования эндоцитоза, проникновения вирусов и их репликации. Более того, SARS-CoV-2 изменяет транскриптом мегакариоцитов через ACE2-независимый, но CD147-зависимый механизм, что может объяснить сообщения о поглощении SARS-CoV-2 тромбоцитами и мегакариоцитами.
Хотя гиперактивность тромбоцитов характерна для COVID-19, ингибиторы тромбоцитов не обеспечили убедительного преимущества в предотвращении полиорганной недостаточности или САС. Например, в рандомизированном исследовании более 500 госпитализированных некритических пациентов с COVID-19 добавление ингибиторов P2Y12 к терапевтическим дозам гепарина не увеличило вероятность отсутствия острого респираторного дистресс-синдрома, не снизило потребность в органной поддержке, не улучшило выживаемость на 21-й день и не уменьшило количество тромботических событий.
Кроме того, в рандомизированном контролируемом исследовании примерно 15 000 пациентов с COVID-19 был сделан аналогичный вывод, что аспирин (который ограничивает функцию тромбоцитов путем ингибирования циклооксигеназы-1) не оказал значительного влияния на частоту механической вентиляции или смертность. Однако в группе, получавшей аспирин, наблюдалось незначительное снижение числа тромботических событий и небольшое увеличение частоты выписки в течение 28 дней.
На многие вопросы оценки вклада гипервоспаления и дисфункции тромбоцитов в развитие САС еще предстоит найти ответ.
Гипервоспаление и коагулопатия при COVID-19: основные пробелы в знаниях
? Какую роль играют нейтрофильные внеклеточные ловушки (NETs) в CAC? Являются ли циркулирующие NETs маркером тяжести заболевания и можно ли безопасно воздействовать на них для улучшения клинических исходов?
? Каковы ключевые продукты активации комплемента и пути, способствующие развитию САС? Одинаковы ли они во всем сосудистом русле?
? Какие пути и компоненты комплемента являются наиболее эффективными и безопасными терапевтическими мишенями?
? Если предположить, что SARS-CoV-2 может инфицировать тромбоциты и мегакариоциты, каковы механизмы проникновения вируса в клетки? Существуют ли специфические вирусные рецепторы, помимо ангиотензин-превращающего фермента 2 (ACE2)? Могут ли модифицированные гликаны участвовать в проникновении вируса?
? Можно ли использовать транскриптомику тромбоцитов в качестве инструмента для прогнозирования риска тромбоза?
? Какие «новые» молекулярные пути, регулирующие активацию тромбоцитов, могут быть направлены на снижение тромбообразования при COVID-19, отдельно или в сочетании с другими?
? Когда в ходе COVID-19 безопасно и наиболее эффективно терапевтически воздействовать на врожденный иммунный ответ, обеспечивая при этом адекватный ответ хозяина на инфекцию?
Гиперкоагуляция при CАС
Многочисленные исследования зафиксировали повышенную прокоагулянтную и подавленную фибринолитическую активность у пациентов с COVID-19, что в совокупности может привести к развитию САС. Исследование, в котором сравнивалась степень этих нарушений у госпитализированных пациентов с COVID-19 и пациентов с сепсисом, выявило повышенный уровень растворимого тромбомодулина в плазме крови в обеих группах, что соответствует повышенной активации эндотелиальных клеток.
В обеих группах пациентов также наблюдалось замедленное, но усиленное образование фибрина и повышенная устойчивость фибрина к фибринолизу. Однако плазма пациентов с COVID-19 и пациентов с сепсисом демонстрировала различные нарушения в кинетике образования тромбина и фибринолитической активности. В то время как плазма пациентов с сепсисом имела нормальный потенциал генерации тромбина, плазма пациентов с COVID-19 имела повышенную скорость генерации тромбина.
Плазма пациентов с сепсисом также показала замедленную выработку плазмина, тогда как у пациентов с COVID-19 потенциал выработки плазмина был нормальным. Наконец, в плазме крови пациентов с COVID-19 наблюдалась задержка образования тромбина, плазмина и фибрина, а также повышение риска по шкале SOFA (Sequential Organ Failure Assessment), служащей для оценки органной недостаточности.
Транскриптомное профилирование БАЛЖ пациентов с COVID-19 по сравнению с лицами без COVID-19 продемонстрировало дифференциальную регуляцию генов, кодирующих коагуляционные и фибринолитические белки. Повышенные транскрипты TF были обнаружены в образцах клеток БАЛЖ от пациентов с COVID-19. SARS-CoV-2 также связан с повышением уровня генов, кодирующих прокоагулянтные белки (например, фактор XI, фактор VII и фактор фон Виллебранда (vWF)), и снижением уровня генов, кодирующих антикоагулянты (тромбомодулин и протеин S) и профибринолитические белки (урокиназа и рецептор активатора плазминогена урокиназы).
Дисрегуляция системы протеина С, способствующая развитию САС
Активированный протеин С — это естественная сериновая протеаза плазмы крови, обладающая как антикоагулянтной активностью, так и прямой клеточной сигнальной активностью, требующей прежде всего EPCR (эндотелиальный рецептор белка С) и протеазно-активируемых рецепторов (PARs) PAR1 и PAR3. Появляется все больше доказательств того, что целостность системы протеина С, необходимой для выработки активированного протеина С, нарушается при САС.
Низкий уровень протеина С у госпитализированных пациентов с СОVID-19 был связан с более тяжелым течением заболевания и повышенной смертностью. Это же отражено в наблюдениях за пациентами с сепсисом и ДВС-синдромом, где низкий уровень протеина С был полезным биомаркером для выявления рисков развития тяжелого заболевания. Низкий уровень протеина С, который повышает риск ишемического инсульта и деменции, может также повышать риск инсульта у пациентов с COVID-19 или «мозгового тумана», одного из признаков LongCOVID.
Повышение уровня растворимого тромбомодулина, который напрямую коррелирует с повреждением эндотелиальных клеток легких и тяжестью заболевания, может указывать на нарушение выработки активированного протеина С. В пользу снижения выработки активированного протеина С, а также повреждения и активации эндотелиальных клеток свидетельствуют и сообщения о повышенных уровнях растворимого EPCR у пациентов с COVID-19. Выделение EPCR из воспаленного эндотелия может не только снизить способность генерировать активированный протеин С, но и уменьшить способность активированного протеина С вызывать «цитопротекторную сигнализацию». Таким образом, вымывание эндотелиального тромбомодулина и EPCR может вызвать дисфункцию пути протеина С, что, в свою очередь, вызывает клеточную резистентность к активированному протеину С и ограничивает его способность восстанавливать баланс дисрегулированных воспалительных и коагулянтных путей.
Система протеина С представляет собой потенциальную мишень для механистических и терапевтических исследований САС. Цитопротекторное действие активированного протеина С и/или его вариантов может быть уникально эффективным для замедления иммунотромбоза при COVID-19, тем более что EPCR-зависимые PAR1 и PAR3 сигнальные пути также вызывают противовоспалительные эффекты.
Антифосфолипидные антитела
Необычной особенностью САС, среди тромботических расстройств в целом, является то, что она включает тромбоз в артериях, венах и микроциркуляторном русле — спектр, напоминающий пациентов с тяжелыми антифосфолипидными синдромами. Действительно, имеются многочисленные сообщения о случаях развития у пациентов с COVID-19 тяжелых тромботических нарушений, сопровождающихся образованием антифосфолипидных антител и волчаночных антикоагулянтов. Однако преходящее появление антифосфолипидных антител может сопровождать и другие вирусные инфекции, поэтому еще предстоит выяснить, действительно ли у пациентов с COVID-19 развивается антифосфолипидный синдром и какую роль он может играть в развитии тромбоза и повреждении органов у этих пациентов.
Интересно, что в некоторых исследованиях сообщалось, что выделенный IgG из сыворотки пациентов с COVID-19 с высоким титром антифосфолипидных антител является сильным протромботическим фактором при введении мышам и способен активировать культивированные эндотелиальные клетки человека. Очевидно, что необходимы дополнительные исследования, чтобы определить, являются ли антифосфолипидные антитела у пациентов с COVID-19 причиной САС, и если да, то сохраняются ли такие антитела у этих пациентов.
Ниже перечислены ключевые вопросы, которые необходимо решить для лучшего понимания гиперкоагуляции, возникающей при COVID-19.
Гиперкоагуляция при COVID-19: нерешенные научные задачи
? Почему диссеминированное внутрисосудистое свертывание крови не является основным признаком COVID-19?
? Можно ли рассматривать комбинированные методы лечения для «восстановления баланса» изменений в коагуляции и фибринолизе при COVID-19, тем самым минимизируя риск развития CAC?
? Каков относительный вклад респираторных эпителиальных клеток в высвобождение коагуляционных, фибринолитических белков и белков комплемента, которые влияют на местное и системное тромбообразование и САС? Можно ли воздействовать на эти клетки для снижения тромботического риска?
? Какие компоненты системы протеина С и его последующие эффекторы являются наилучшими кандидатами в качестве терапевтических мишеней для профилактики и/или лечения САС? Можно ли рассматривать комбинированные методы лечения?
? Каковы механизмы, посредством которых антифосфолипидные антитела могут способствовать развитию САС?
Заключение
Хотя за годы пандемии многое стало известно о патофизиологии COVID-19, понимание пусковых событий COVID-19-ассоциированной коагулопатии остается непростым, как и выработка эффективных профилактических и терапевтических стратегий. Это неудивительно, учитывая, что САС — очень сложный и динамичный процесс, включающий взаимодействие множества биохимических, протеолитических и клеточных путей. Дифференциация того, какие механизмы являются причинно-следственными, последовательными или просто случайными, крайне затруднена.
Отсюда и проблема определения, на какой путь следует воздействовать терапевтически и когда это делать в ходе болезни, не нарушая естественных защитных механизмов. Вполне вероятно, что один или несколько механизмов САС преобладают на разных стадиях заболевания, в разных органах и сосудистых руслах. В этом отношении мы лучше представляем себе ранние стадии COVID-19 после инфицирования, когда вирус повреждает альвеолы и прилегающие микрососуды с первоначальным местным гипоксическим и иммунотромботическим ответом. При последующем развитии заболевания выявление ключевых триггеров в сложной иммунотромботической сети становится все более сложной задачей.
Поэтому по-прежнему актуально продолжать практические направления исследований, которые могут принести клиническую пользу. В условиях сохраняющейся неопределенности с появлением новых вариантов SARS-CoV-2 такие целенаправленные исследования имеют решающее значение для разработки эффективных антииммунотромботических профилактических и терапевтических стратегий для пациентов с коронавирусной инфекцией.